Run the MEGAHIT assembler¶
MEGAHIT is a very fast, quite good assembler designed for metagenomes.
First, install it:
cd ~/
git clone https://github.com/voutcn/megahit.git
cd megahit
make
Now, download some data:
mkdir ~/data
cd ~/data
curl -O https://s3-us-west-1.amazonaws.com/dib-training.ucdavis.edu/metagenomics-scripps-2016-10-12/SRR1976948.abundtrim.subset.pe.fq.gz
curl -O https://s3-us-west-1.amazonaws.com/dib-training.ucdavis.edu/metagenomics-scripps-2016-10-12/SRR1977249.abundtrim.subset.pe.fq.gz
These are data that have been run through k-mer abundance trimming (see k-mer and subsampled so that we can run an assembly in a fairly short time period).
Now, finally, run the assembler!
mkdir ~/assembly
cd ~/assembly
ln -fs ../data/*.subset.pe.fq.gz .
~/megahit/megahit --12 SRR1976948.abundtrim.subset.pe.fq.gz,SRR1977249.abundtrim.subset.pe.fq.gz \
-o combined
This will take about 15 minutes; at the end you should see output like this:
... 7713 contigs, total 13168567 bp, min 200 bp, max 54372 bp, avg 1707 bp, N50 4305 bp
... ALL DONE. Time elapsed: 899.612093 seconds
The output assembly will be in combined/final.contigs.fa.
While the assembly runs...¶
- Discuss CAMI paper.
- What does, and doesn’t, assemble?
- How good is assembly anyway?
Questions for Discussion:¶
- Why would we assemble, vs looking at raw reads? What are the advantages and disadvantages?
- What are the technology tradeoffs between Illumina HiSeq, Illumina MiSeq, and PacBio? (Also see [this paper (http://ivory.idyll.org/blog/2015-sharon-paper.html).)
- What kind of experimental design considerations should you have if you plan to assemble?
Some figures for your consideration: The first two come from work by Dr. Sherine Awad on analyzing the data from Shakya et al (2014). The third comes from an analysis of read search vs contig search of a protein database.
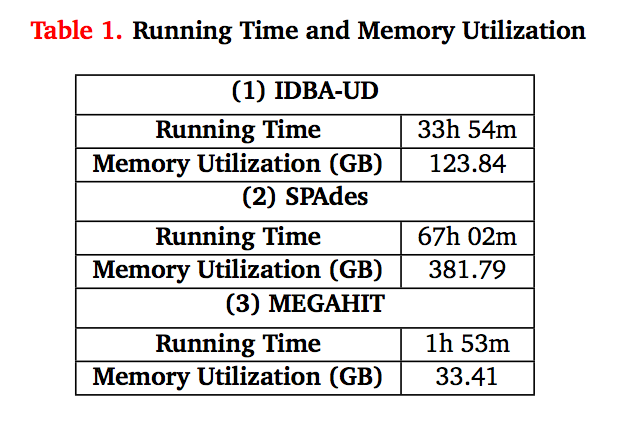
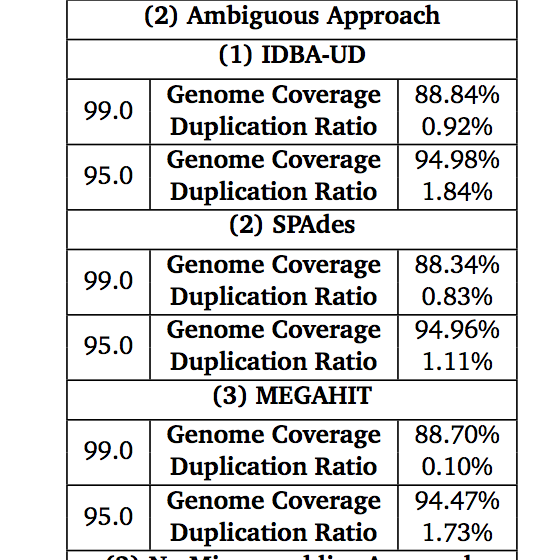
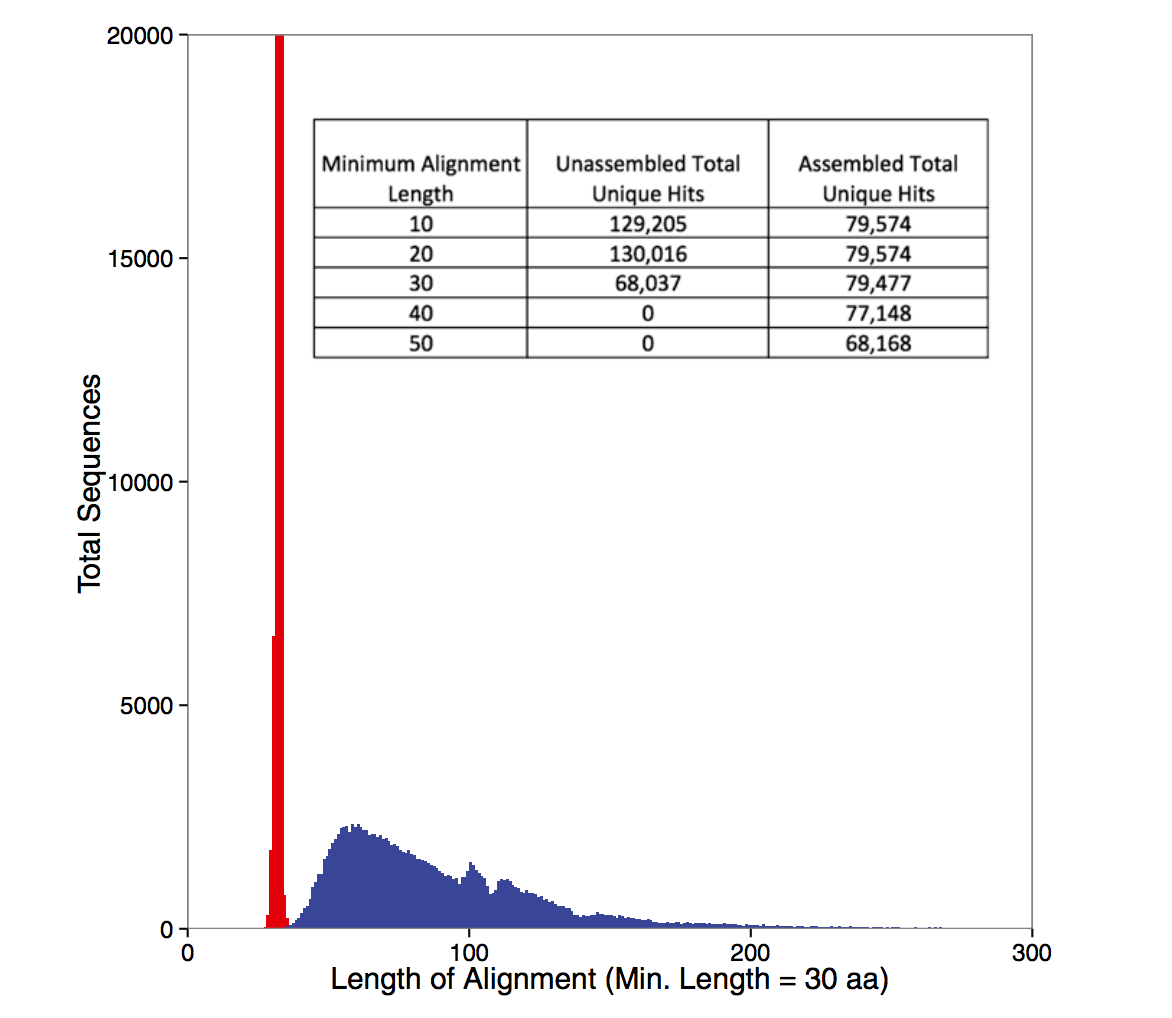
After the assembly is finished¶
Let’s first take a look at the assembly:
less combined/final.contigs.fa
Now we can run a few stats on our assembly. To do this we will use QUAST:
cd ~/
git clone https://github.com/ablab/quast.git -b release_4.5
export PYTHONPATH=$(pwd)/quast/libs/
Now, run QUAST on the assembly:
cd ~/assembly
~/quast/quast.py combined/final.contigs.fa -o combined-report
cat combined-report/report.txt
What does this say about our assembly? What do the stats not tell us? For thought and discussion: a few nice slides from Jessica Blanton on assessing assembly quality.
At this point we can do a bunch of things:
- annotate the assembly with Prokka;
- evaluate the assembly’s inclusion of k-mers and reads;
- set up a BLAST database so that we can search it for genes of interest;
- quantify the abundance of the contigs or genes in the assembly, using the original read data set Salmon;
- Identify contigs with similar tetra-mer abundance and coverage with Binning;
LICENSE: This documentation and all textual/graphic site content is released under Creative Commons - 0 (CC0) -- fork @ github.